Docking simulation for Drug discovery using Grid Computing
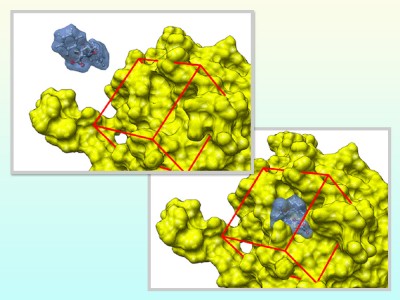
Docking simulation investigates whether a chemical compound and a protein can be bonded or not. In practical situation, a number of combination of them are investigated. In this study, we aim to contribute to this scientific field as well as streamline docking simulation by distributing the computational workloads to computational resources on the Grid environment. This research has been performed on the Grid testbed by PRAGMA research community in which more than 30 institutions and universities have participated.
Most of reserch products and achivements are produced by graduate and undergraduate students in Osaka Universitya and UCSD.
Research achievement
- Jason H. Haga, Kohei Ichikawa, Susumu Date, “Virtual Screening Techniques and Current Computational Infrastructures”, Current Pharmaceutical Design, Vol.22, No. 23, pp. 3576-3584, 2016. [DOI: 10.2174/1381612822666160414142530]
- Daniel Li, Brian Tsui, Charles Xue, Jason H. Haga, Kohei Ichikawa, Susumu Date, “Protein Structure modeling in a Grid computing environment”, Proceedings of The 9th IEEE International Conference on e-Science (e-science 2013), pp. 301-306, Beijing, China, Oct. 2013.
- Wen-Wai Yim, Shu Chien, Yasuyuki Kusumoto, Susumu Date and Jason Haga, “Grid Heterogeneity in In-silico Experiments: An Exploration of Drug Screening Using DOCK on Cloud Environments”, Healthgrid Applications and Core Technologies (Proceedings of HealthGrid 2010), IOS Press, pp.181-190, Paris, June 2010.
- Marshall J. Levesque, Kohei Ichikawa, Susumu Date and Jason Haga, “Design of a Grid-Service-based Platform for in silico Protein-Ligand Screenings”, Computer Methods and Programs in Biomedicine, Vol. 93, No. 1, pp. 73-82, Jan. 2009.
Related Theme
